LRRK2 BAC transgenic rats develop progressive, L-DOPA-responsive motor impairment, and deficits in dopamine circuit function.
Changes in the LRRK2 gene are one of the most common genetic contributors to Parkinson’s disease. To better understand how changes in LRRK2 affects nerve cells, we studied rats expressing types of LRRK2 genes found in patients. We show these LRRK2 variants lead to movement difficulties and alter the electrical and chemical signals of the dopamine-releasing nerve cells vulnerable in Parkinson’s.
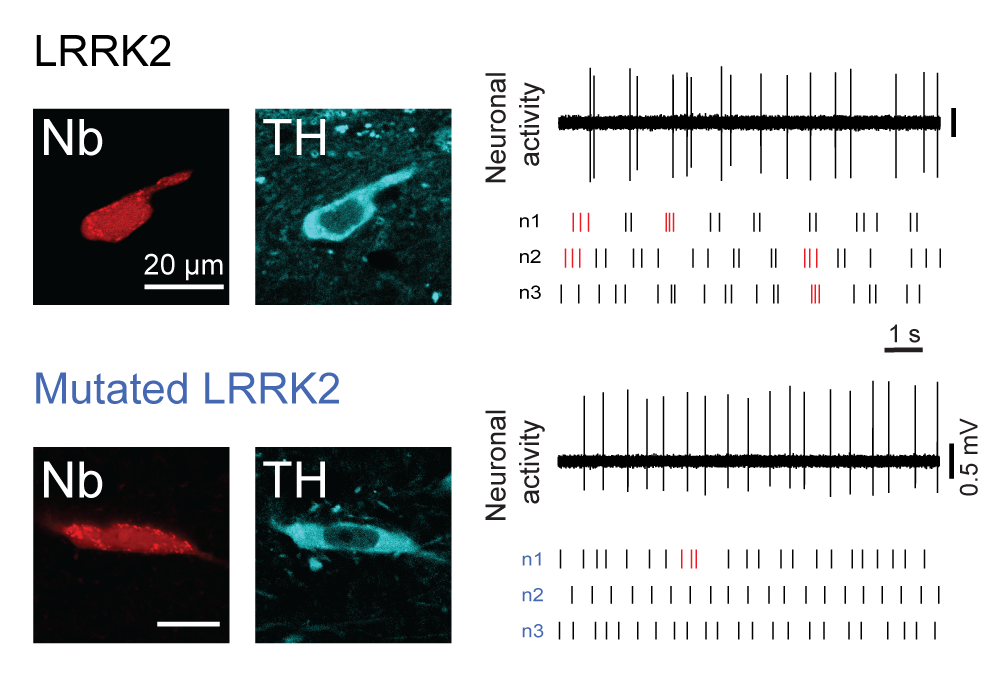
Mutations in leucine-rich repeat kinase 2 (LRRK2) lead to late-onset, autosomal dominant Parkinson's disease, characterized by the degeneration of dopamine neurons of the substantia nigra pars compacta, a deficit in dopamine neurotransmission and the development of motor and non-motor symptoms. The most prevalent Parkinson's disease LRRK2 mutations are located in the kinase (G2019S) and GTPase (R1441C) encoding domains of LRRK2. To better understand the sequence of events that lead to progressive neurophysiological deficits in vulnerable neurons and circuits in Parkinson's disease, we have generated LRRK2 bacterial artificial chromosome transgenic rats expressing either G2019S or R1441C mutant, or wild-type LRRK2, from the complete human LRRK2 genomic locus, including endogenous promoter and regulatory regions. Aged (18-21 months) G2019S and R1441C mutant transgenic rats exhibit L-DOPA-responsive motor dysfunction, impaired striatal dopamine release as determined by fast-scan cyclic voltammetry, and cognitive deficits. In addition, in vivo recordings of identified substantia nigra pars compacta dopamine neurons in R1441C LRRK2 transgenic rats reveal an age-dependent reduction in burst firing, which likely results in further reductions to striatal dopamine release. These alterations to dopamine circuit function occur in the absence of neurodegeneration or abnormal protein accumulation within the substantia nigra pars compacta, suggesting that nigrostriatal dopamine dysfunction precedes detectable protein aggregation and cell death in the development of Parkinson's disease. In conclusion, our longitudinal deep-phenotyping provides novel insights into how the genetic burden arising from human mutant LRRK2 manifests as early pathophysiological changes to dopamine circuit function and highlights a potential model for testing Parkinson's therapeutics.
2024. Nat Metab, 6(11):2100-2117.
